Afrika Trypanosomiasisi olarak da bilinen Afrika Uyku Hastalığı, farklı aşamalardan geçen tropikal bir hastalıktır. Sonunda meningoensefalit, koma ve ölümle sonuçlanır. Bu hastalık, Sahra altı Afrika’da 60 ila 70 milyon insanı etkilemektedir.
Haber Merkezi / Dünya Sağlık Örgütü (DSÖ), hastalığın Afrika’nın birçok bölgesinde önemli bir ölüm nedeni olduğunu bildirmektedir.
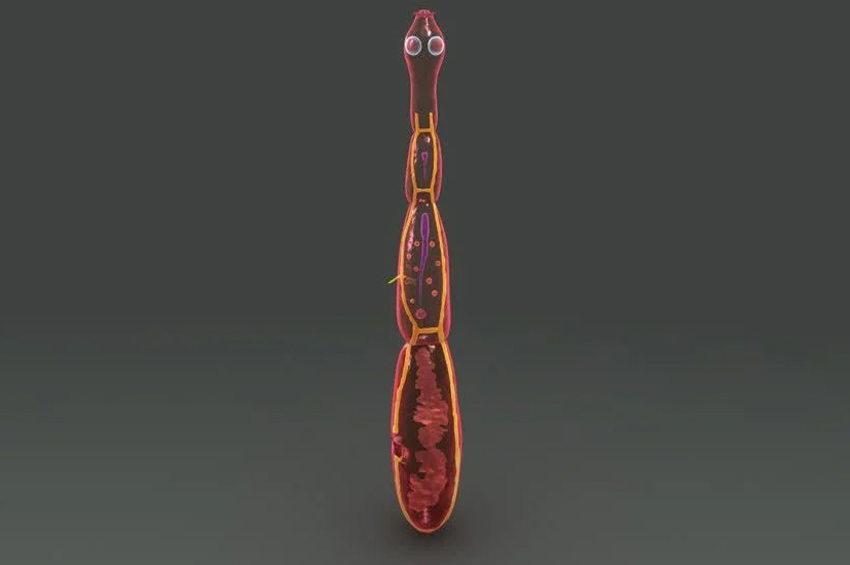
Hastalığa, enfekte çeçe sineklerinin ısırması ile yayılan Trypanosoma brucei parazitleri neden olur. Bulaşması öncelikle enfekte bir çeçe sineğinin tek bir ısırması yoluyla olur. Parazit insanları ve diğer memelileri enfekte edebilir.
Hastalığın erken dönemlerinde Trypanosoma brucei parazitleri kan, lenf ve deri altı dokularında yaşar. Bu aşamada hastalar baş ağrısı, eklem ağrısı ve ateş yaşarlar. Tedavi edilmezse parazit daha sonra merkezi sinir sistemine (CNS) girer. CNS’yi istila ettiklerinde, parazitler davranış değişikliklerine, kafa karışıklığı gibi bilişsel işlev bozukluklarına ve önemli uyku bozukluklarına neden olur.
Trypanosoma brucei paraziti , çeçe sineği vektörü tarafından bulaşmaya ek olarak , parazit plasentayı istila ettiğinde anneden çocuğa dikey olarak bulaşabilir. Mekanik bulaşma ayrıca diğer kan emici böcekler (örneğin sivrisinekler), sağlık bakım ortamlarında iğne batması yaralanmalarından kaynaklanan kaza sonucu aşılama veya cinsel yolla bulaşma yoluyla da gerçekleşebilir. Bununla birlikte, bu enfeksiyon yolları nadirdir.
Kimler risk altında?
Afrika Uyku Hastalığı’nın iki türü vardır. Her birine farklı bir Trypanosoma brucei paraziti neden olur:
- Trypanosoma brucei rhodesiense: Doğu Afrika’da öne çıkan bu hastalık türü, enfeksiyonların %2’sinden daha azından sorumludur. Hastalık, enfeksiyondan sonraki birkaç hafta içinde hızlı semptom gelişimi ile karakterizedir. Bu daha şiddetli Afrika Uyku Hastalığı türünde parazitler merkezi sinir sistemini kolaylıkla istila eder ve aylar içinde ölüm meydana gelir.
- Trypanosoma brucei gambiense: Batı Afrika’da öne çıkan bu hastalık türü, vakaların %98’inden sorumludur. İlk semptomlar, diğer tıbbi durumlarla ilişkili olabilecek durumlar olan aralıklı ateş ve baş ağrılarını içerir. Bu göz önüne alındığında, Afrika Uyku Hastalığı teşhisinde zorluk vardır. Tanı genellikle parazit merkezi sinir sistemine ulaştığında ve hastalığın temel özellikleri kendini gösterdiğinde konur.
Afrika Uyku Hastalığı, Afrika’nın birçok bölgesinde görüldüğünden, yalnızca kıtaya seyahat edenler veya orada yaşayanlar, durumu edinme açısından birincil risk altındadır. Tarım, avcılık veya balıkçılığın birincil faaliyet olduğu kırsal Afrika köylerinde enfeksiyon riski artmıştır. Afrika ülkeleri arasında, durum için en fazla risk altındaki ülkeler Çad, Kongo, Angola, Malavi, Tanzanya, Sudan, Zambiya ve Uganda’dır.

Parazit gelişiminin aşamaları
Trypanosoma brucei parazitinin 2 alt türü , Afrika Uyku Hastalığının farklı belirtilerine neden olur. Parazit gelişimi, çeçe sineği bir memeli konakçı (örneğin insan) ile beslendiğinde başlar ve metasiklik tripomastigotlar vücut dokularına enjekte edilir. Parazit lenf sistemine ve kan dolaşımına girdiğinde, parazit 3 hafta boyunca birkaç aşamadan geçer:
- Parazitler kan dolaşımı tripomastigotlarına dönüşür.
- Daha sonra vücut sıvılarını işgal ettikleri ve ikili bölünme yoluyla çoğaldıkları başka yerlere taşınırlar.
- Daha uzun enfeksiyon süreleri, parazitin Afrika Uyku Hastalığı’nın temel semptomlarına neden olduğu merkezi sinir sisteminin parazit istilası ile ilişkilidir.
Belirtileri
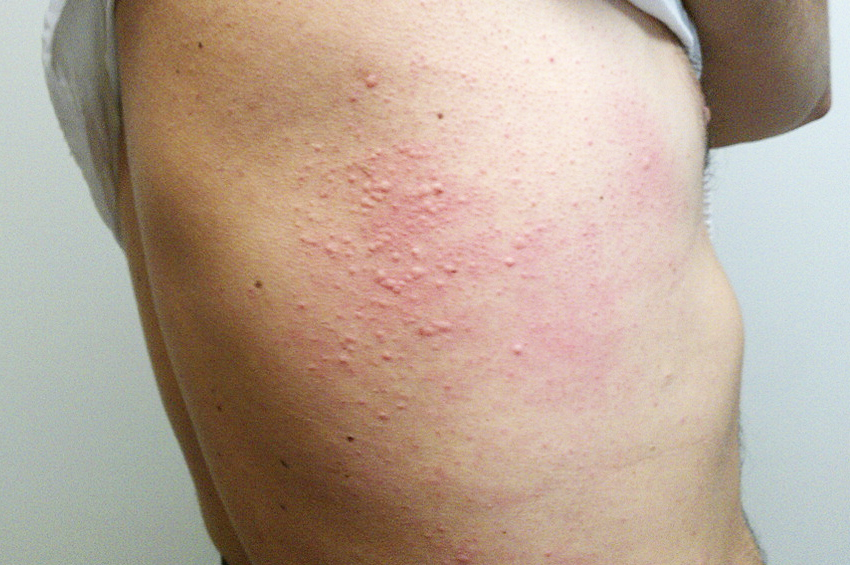
Afrika uyku hastalığı tedavi edilmediğinde yüksek morbiditeye neden olan bir durumdur. Doğu Afrika uyku hastalığının en erken belirtisi, bir çeçe sineği tarafından ısırıldıktan yaklaşık beş gün sonra ortaya çıkan Trypanosomal şans adı verilen yerel bir cilt reaksiyonudur.
Trypanosoma brucei ile genel bir enfeksiyonun ilk ve en yaygın tezahürü , vücutta parazitik çoğalma ile uyumlu periyodik bir döngü içinde gelen ve giden ateşin ortaya çıkmasıdır. Diğer semptomlar baş ağrısı, kas ağrısı ve eklem ağrısıdır.
Doğu Afrika uyku hastalığında, bu klinik özellikler enfeksiyondan sonraki birkaç hafta içinde ortaya çıkar ve lenfadenopatiyi de içerebilir. Bu tür uyku hastalığı daha şiddetlidir ve birkaç hafta içinde miyokardiyal disfonksiyon ve ölümle sonuçlanabilir.
Batı Afrika tripanozomiyazı, hepatosplenomegali ve döküntü gibi daha az belirgin semptomlarla kendini gösterir ve erken teşhisi zorlaştırır. Bir kişiye Batı Afrika Uyku Hastalığı bulaşmasından birkaç ay sonra, parazit kan-beyin bariyerini (BBB) geçmeye ve merkezi sinir sistemine (CNS) girmeye başlar.
Bu aşamada hastalar, kronik ensefalopatinin neden olduğu zihinsel ve bilişsel değişiklikleri yaşamaya başlar. Bu tipik olarak, hastanın uyku düzeninin bozulmasıyla birlikte uzun süre konsantre olamaması olarak kendini gösterir.
Tedavisi
Afrika Uyku Hastalığı teşhisi konan ve erken ve spesifik tedavi gören hastaların hayatta kalma şansları çok yüksektir. Bununla birlikte, bu hastalığın erken evrelerinde tanımlanamayan semptomlar nedeniyle, genellikle geç teşhis edilir.
Tedavi planı, hastalığın klinik görünümüne ve paraziter nedene bağlıdır; Vakaların ~%2’sine neden olan ve daha şiddetli olan Trypanosoma brucei rhodesiense (Doğu Afrika Uyku Hastalığı) veya ~%98 hastayı etkileyen ve kronik bir hastalık olan Trypanosoma brucei gambiense (Batı ve Orta Afrika uyku hastalığı).
Aşağıda, Afrika Uyku Hastalığı tedavisinde başlıca kullanılan beş kimyasal madde yer almaktadır:
1 aşama tedavisi için:
- Pentamidin isethionate; Bu kimyasal bileşik 1940 yılında keşfedilmiştir ve öncelikle Trypanosoma brucei gambiense (Batı Afrika uyku hastalığı) ile enfekte olan hastaları tedavi etmek için kullanılır.
- Sodyum suramin; Bu kimyasal bileşik 1920’de keşfedildi ve öncelikle Trypanosoma brucei rhodesiense (Doğu Afrika Uyku Hastalığı) ile enfekte olan hastaları tedavi etmek için kullanılıyor.
2 aşama tedavisi için:
- Arsenik türevleri; Bu bileşikler, Afrika uyku hastalığının her iki tipini tedavi etmek için kullanılabilir ve Trypanosoma brucei rhodesiense için birinci basamak tedavi ve dirençli Trypanosoma brucei gambiense için ikinci seçenek olarak kabul edilir . Bununla birlikte, son araştırmalar Orta Afrika’daki çeşitli popülasyonlarda dirençli T. brucei gambiense suşlarının oldukça yaygın olduğunu göstermiştir.
- Alfa-diflorometilornitin: Trypanosoma brucei gambiense tedavisinde etkili olmakla birlikte , bu bileşik daha karmaşık ve titiz bir tedavi planı gerektirir ve bu nedenle nadiren kullanılır.
- Nitrofuran tiyazin: Alfa-diflorometilornitin ve nitrofuran tiyazini birleştiren ve 2009’da geliştirilen bir tedavi planı. Bu tedavi daha az sıklıkta tedavi gerektirir ve bu nedenle DSÖ tarafından T. gambiense enfeksiyonu için birinci basamak tedavi olarak önerilmektedir.
Afrika uyku hastalığı için sınırlı sayıda farmakolojik müdahale vardır. Bu durumla ilgili daha fazla araştırmanın daha etkili tedavi ile sonuçlanacağı umulmaktadır.
Prognoz
Erken evrelerinde, Afrika Uyku Hastalığının semptomları diğer birçok durumdan farklı değildir, bu da bazı hastaların kronik evreye girene kadar teşhis edilmeyeceği anlamına gelir. Akut faz çok hızlı gerçekleştiği için erken teşhis önemlidir.
Bu hastalık, enfeksiyondan yaklaşık iki ila üç hafta sonra, parazit CNS’ye ulaşmadan önce teşhis edilirse tedavi edilebilir. Evre 1 tedavisinin bir yan etkisi olarak ensefalopati geliştirecek tahmini %2.5’i dışında, bu süre içinde uygun tedaviyi alan hastaların neredeyse tamamı tamamen iyileşir.
Afrika uyku hastalığının prognozu ile ilgili mevcut çalışmalar, ikinci aşamada tedavi hala mümkün olsa da, hayatta kalma oranının düşük olduğunu göstermektedir. BBB’nin parazit tarafından geçmesi nedeniyle enfeksiyondan sonraki altı ay boyunca tedavi edilmeyen hastalarda mortalite de yüksektir.
Hasta ölüm kayıtları, Afrika uyku hastalığından ölümlerin %80’inden fazlasının, duruma çok az müdahale edilmesi veya hiç müdahale edilmemesi nedeniyle meydana geldiğini göstermektedir.
Dikkat: Sayfa içeriği sadece bilgilendirme amaçlıdır.