VHL veya von Hippel-Lindau hastalığı, VHL genindeki bir silme veya mutasyondan kaynaklanan otozomal dominant bir genetik durumdur. VHL hastalığı ortalama 36.000 kişiden 1’ini etkiler ve hastaların %20’si ailede ilk veya yeni vakalardır.
Haber Merkezi /Ortalama başlangıç yaşı 26’dır ve VHL gen mutasyonu olan kişilerin %97’sinde 65 yaşına kadar semptomlar görülür. VHL hastalığı erkekleri, kadınları ve tüm etnik grupları eşit şekilde etkiler ve dünyanın her yerinde görülür.
VHL hastalığı olan kişiler, beyin, omurga, gözler, böbrekler, pankreas, adrenal bezler, iç kulaklar, üreme sistemi, karaciğer ve akciğer dahil olmak üzere vücudun on kadar yerinde tümörler ve/veya kistlerle karşılaşabilir:
Belirtileri ve semptomları
VHL hastalığının tek bir birincil semptomu yoktur. Bunun nedeni kısmen, yalnızca vücudun bir organında oluşmamasıdır. Ayrıca her zaman belirli bir yaş grubunda görülmez. Durum kalıtsaldır, ancak aynı genetik mutasyona rağmen hastalığın sunumu bireyler arasında çok farklı olabilir. Ek olarak, VHL lezyonlarının görünümü ve ciddiyeti insanlar arasında o kadar farklıdır ki, aynı ailenin birçok üyesinde yalnızca nispeten zararsız bir sorun olabilirken, diğerlerinde ciddi bir hastalık olabilir.
Başlangıç yaşı aileden aileye ve bireyden bireye değişir. Bazı ailelerde feokromositomalar (adrenal tümörler) çok yaygınken, diğer ailelerde berrak hücreli renal hücreli karsinomlar (böbrek tümörleri) daha yaygındır.
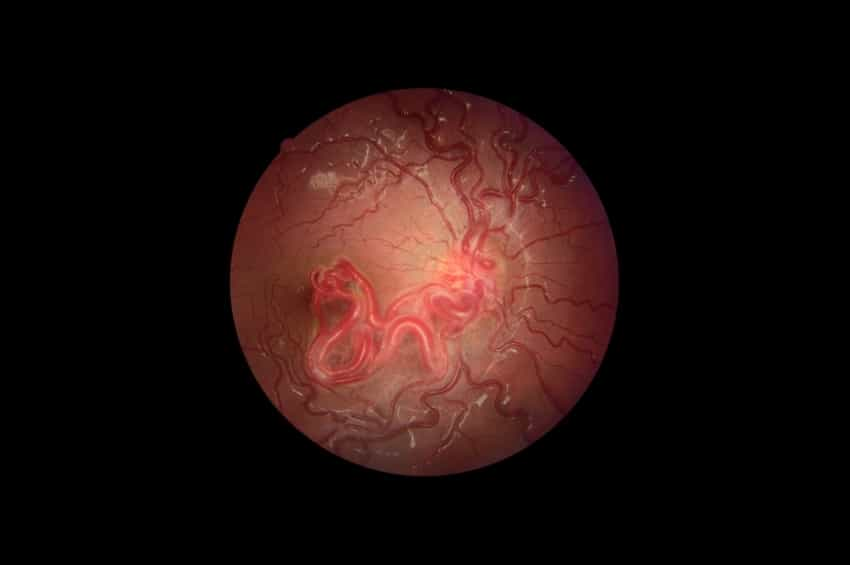
VHL’nin en yaygın semptomu hemanjiyoblastomlardır. Bunlar beyin, omurilik ve retinada meydana gelen iyi huylu tümörlerdir. Hemanjiyoblastomlar iyi huyludur. Beyinde veya omurilikte, hemanjiyoblastom bazı durumlarda bir kist veya sıvı dolu kese içinde bulunabilir. Hemanjiyoblastomlar veya çevredeki kistler sinir veya beyin dokusuna baskı yaparak baş ağrısı, yürürken denge sorunları veya kol ve bacaklarda güçsüzlük gibi semptomlara neden olabilir. Gözlerde, hemanjiyoblastomlardan kan veya sıvı sızıntısı görmeyi engelleyebilir. Erken teşhis, gözlerin dikkatli bir şekilde izlenmesi ve hızlı tedavi, sağlıklı görmeyi sürdürmek için çok önemlidir.
Adrenal tümörlerin erken belirtileri yüksek tansiyon, panik ataklar veya aşırı terleme olabilir. Pankreas kistlerinin ve tümörlerinin erken belirtileri, şişkinlik veya bağırsak ve mesane fonksiyonlarının bozulması gibi sindirim şikayetlerini içerebilir. Bu tümörlerin bazıları iyi huylu iken bazıları kansere dönüşebilir.
Böbrek tümörleri ve kistler (berrak hücreli renal hücreli karsinom) böbrek fonksiyonlarında azalmaya yol açabilir, ancak genellikle erken evrelerde hiçbir belirti görülmez. Böbrek tümörleri, yaklaşık 3 cm çapa ulaştıklarında çıkarılmadıkları takdirde metastaz yapacaklardır.
VHL ayrıca iç kulakta endolenfatik kese tümörü adı verilen iyi huylu bir tümöre neden olabilir. Bu tümör çıkarılmazsa etkilenen kulakta işitme kaybına ve denge sorunlarına yol açabilir. VHL’nin daha az görülen belirtileri, hem erkeklerde hem de kadınlarda iyi huylu üreme sistemi tümörlerini içerir. Bununla birlikte, bu tümörler hamile kalma sorunlarına veya hamile kalmaya yol açabilir. Karaciğer ve akciğerdeki tümörler problemsiz kabul edilir.
Nedenleri
VHL hastalığı, 3. kromozomun kısa kolunda bulunan VHL genindeki bir delesyon veya mutasyondan kaynaklanan otozomal dominant bir hastalıktır. VHL’li bir kişinin her çocuğu, genin değiştirilmiş kopyasını kalıtım yoluyla alma konusunda %50 risk altındadır.
Normal VHL geni, tümör oluşumunu önleme işlevi ile bir tümör baskılayıcı gen görevi görür. Gen, ürünü olan VHL proteini (pVHL) aracılığıyla hücresel hipoksi sinyalinin ana düzenleyicisi olarak görev yapar. HIF (hipoksi-indüklenebilir faktör) kompleksi yoluyla pVHL, vasküler endotelyal faktör, trombositten türetilmiş büyüme faktörü ve dönüştürücü büyüme faktörü alfa dahil olmak üzere artan büyüme faktörlerinden dolaylı olarak sorumludur.
VHL hastalığında olduğu gibi işlev görmeyen bir gen durumunda, HIF kompleksinin düzenlenmesi gerçekleşmez. Sonuç, artan kan damarı büyümesine (anjiyogenez) ve tümör oluşumuna izin veren çeşitli büyüme faktörlerinin artan seviyeleridir. Bu, böbrek, meme, pankreas, adrenal kanserler gibi diğer daha yaygın kanserlerde yer alan aynı süreçtir.
Araştırmacılar, büyüme faktörü ve/veya HIF aktivitesine müdahale etmenin VHL ve diğer kanser türleri için etkili bir tedavi olacağını kanıtlayacağına inanıyor.

Teşhisi
VHL’li bir ebeveyni olan herkesin ve VHL’li erkek veya kız kardeşi olan çoğu kişinin VHL hastalığına yakalanma olasılığı %50’dir. VHL’li teyzesi, amcası, kuzeni veya büyükanne ve büyükbabası olan herkes de risk altında olabilir. Birinin değiştirilmiş bir VHL genine sahip olmadığından emin olmanın tek yolu DNA testidir. Bir kişi VHL’ye özgü bir tümör sergilediğinde de klinik tanı konulabilir.
Bir VHL teşhisi konulduktan sonra, herhangi bir semptom ortaya çıkmadan önce sürveyans testine erken başlamak önemlidir. Çoğu VHL lezyonu, küçük olduklarında tedavi edilmesi çok daha kolaydır. VHL’nin bazı olası komplikasyonları, sorun kritik bir düzeye ulaşana kadar semptomlarla kendini göstermez. Tedavi yalnızca meydana gelen semptomları durdurabilir; değişiklikleri tersine çevirmek ve normale dönmek her zaman mümkün değildir.
Tedavisi
Evrensel bir tedavi önerisi mevcut değildir. Tedavi seçenekleri yalnızca her bir hastanın toplam durumunun (semptomlar, test sonuçları, görüntüleme çalışmaları ve genel fiziksel durum) dikkatli bir şekilde değerlendirilmesiyle belirlenebilir. Aşağıdakiler, olası tedavi terapileri için genel kılavuzlar olarak sunulmaktadır.
Beyin ve omurilik hemanjiyoblastomları
Beyin ve omurilikteki hemanjiyoblastomlarla ilgili semptomlar, tümörün yeri, boyutu ve ilişkili şişlik veya kistlerin varlığına bağlıdır. Semptomatik lezyonlar asemptomatik lezyonlardan daha hızlı büyür. Kistler genellikle tümörün kendisinden daha fazla belirtiye neden olur. Lezyon çıkarıldıktan sonra kist çökecektir. Tümörün herhangi bir kısmı yerinde kalırsa, kist yeniden dolar. Semptomatik olmayan ve bir kist ile ilişkili olmayan küçük hemanjiyoblastomlar bazen stereotaktik radyocerrahi ile tedavi edilmiştir, ancak bu bir tedaviden daha önleyicidir ve uzun vadeli sonuçlar yalnızca marjinal fayda gösteriyor gibi görünmektedir. Ayrıca iyileşme döneminde semptomlar azalmayabilir.
Pankreatik nöroendokrin tümörler
Seröz kistadenomları ve pankreas nöroendokrin tümörlerini (pankreatik NET’ler) ayırt etmek için dikkatli analiz gereklidir. Kistler ve kistadenomlar genellikle tedavi gerektirmez. Pankreas NET’leri boyut, davranış ve spesifik genetik mutasyona göre derecelendirilmelidir.
Renal hücreli karsinom
VHL böbrek tümörleri genellikle çok küçük olduklarında ve gelişimin çok erken aşamalarında bulunurlar. Bir bireyin yaşamı boyunca yeterli işleyen bir böbreğe sahip olmasını sağlamaya yönelik bir strateji, yalnızca tümör boyutu veya hızlı büyüme oranı, tümörün metastatik potansiyel kazanabileceğini (yaklaşık 3 cm’de) gösterdiğinde dikkatli izleme ve ameliyatı seçme ile başlar. Böbrek koruyucu cerrahi tekniği bu ortamda yaygın olarak kullanılmaktadır. Özellikle erken evrelerdeki daha küçük tümörler için radyo frekansı ablasyonu (RFA) veya kriyocerrahi (kriyoterapi) düşünülebilir. Komşu yapılara zarar vermemek ve sonraki ameliyatları zorlaştırabilecek yara izini sınırlamak için özen gösterilmelidir.
Retinal hemanjiyoblastomlar
Küçük periferik lezyonlar, lazer kullanılarak çok az veya hiç görme kaybı olmaksızın başarılı bir şekilde tedavi edilebilir. Daha büyük lezyonlar genellikle kriyoterapi gerektirir. Hemanjiyoblastom optik disk üzerindeyse, görüşü başarılı bir şekilde koruyacak birkaç tedavi seçeneği vardır.
Feokromositomalar
İlaçla yeterli blokaj yapıldıktan sonra cerrahi olarak çıkartılır ve laparoskopik parsiyel adrenalektomi tercih edilir. Ameliyattan sonra en az bir hafta boyunca vücut “yeni normale” uyum sağlarken hayati belirtiler dikkatle izlenir. Her türlü cerrahi prosedür sırasında ve hamilelik ve doğum sırasında özel dikkat gösterilmelidir. Aktif görünmeyen veya semptomlara neden olmayan feokromositomalar bile, ideal olarak hamilelikten veya acil olmayan cerrahiden önce çıkarılmalıdır.
Endolenfatik kese tümörleri (ELST’ler)
MRI’da görülebilen bir tümörü veya kanaması olan ancak yine de duyabilen hastaların durumlarının kötüleşmesini önlemek için ameliyat gerekir. Tümörün görüntülenmesiyle ilgili kanıtları olan sağır hastalar, denge sorunlarının kötüleşmesini önlemek için başka nörolojik semptomlar varsa ameliyat edilmelidir. Tüm ELST’ler görüntüleme ile görülmez; bazıları sadece ameliyat sırasında bulunur.
2021’de belzutifan (welireg), ilişkili renal hücreli karsinom, merkezi sinir sistemi hemanjiyoblastomları veya acil ameliyat gerektirmeyen pankreas nöroendokrin tümörleri için tedavi gerektiren von Hippel-Lindau hastalığı olan yetişkin hastalar için onaylandı.