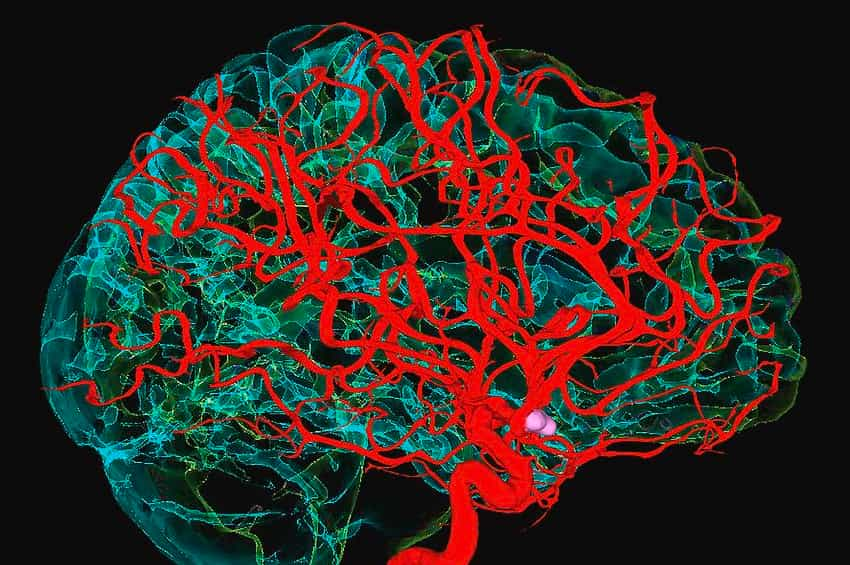
Adından da anlaşılacağı gibi, beynin vasküler malformasyonları (Vascular Malformations of the Brain – VMB), beynin kan damarlarının etkilendiği en az altı durumu kapsayan bir şemsiye terimdir. Bu tür malformasyonlar, semptomların, şiddetinin ve nedenlerinin değiştiği çeşitli tiplerde sınıflandırılır.
Haber Merkezi / Bu VMB türleri şunlardır: (1) arteriovenöz malformasyonlar (AVM), anormal arterler ve damarlar; (2) kavernöz malformasyonlar (CM), genişlemiş kanla dolu boşluklar; (3) venöz anjiyomlar (VA), anormal damarlar; (4) telenjiektaziler (TA), kılcal damar boyutunda genişlemiş damarlar; (5) Galen malformasyonlarının damarı (VGM); ve (6) karışık malformasyonlar (MM).
Belirtileri ve semptomları
Beynin vasküler malformasyonları beyinde baş ağrısı, nöbet, felç veya kanamaya (beyin kanaması) neden olabilir. Bazı araştırmacılar malformasyon tipinin hastalığın semptomlarını ve ilerlemesini belirlediğine inanmaktadır. Diğer araştırmacılar, malformasyonun türünden çok yalnızca ciddiyetinin önemli olduğuna inanıyor.
Arteriyovenöz malformasyonlar veya AVM’ler arterleri, venleri ve orta büyüklükteki damarları etkiler, ancak kılcal damarları etkilemez. Bu kan damarları genişler, bükülür ve birbirine dolanır. Arterler ve toplardamarlar ince kılcal damarlar yerine direkt olarak bağlanabilirler ve bu nedenle kılcal damarlar by-pass edildiğinden sıklıkla “şant lezyonları” olarak adlandırılırlar. Bu anormal “besleyici” arterler giderek genişler ve sonuç olarak “drenaj yapan” damarlar da genişler. Bu damarlar arasındaki beyin dokusu sertleşmiş veya rijit (körelmiş), ince küçük lifler (fibriller) ve düzleştirilmiş hücreler (gliotik) serpiştirilmiş bir ağ ile dolu olabilir ve bazen kireçlenmiş olabilir. Bu tür malformasyonlar kanı beyinden çekerek beyin hücresi atrofisine neden olabilir. AVM’lerde genellikle kanamalar veya nöbetler yaşanır.
Kavernöz malformasyonlar, CM’ler (kavernöz anjiyomlar veya kavernöz hemanjiyomlar veya kavernomlar olarak da adlandırılır), kanla dolu boşlukların anormal şekilde genişlemiş koleksiyonları olarak ortaya çıkar. Kavernöz hemanjiyom, kılcal damarlar arasında, dokular arasındaki boşluklarda (sinüzoidler) ve “daha büyük kavernöz boşluklarda” yolunu bulan kanı emen bir “kan süngeri” gibi davranır. Bunlar “yavaş akışlı lezyonlardır”. AVM’lerin semptomlarının aksine bu boşluklarda genellikle beyin dokusu yoktur. Kanamalar veya nöbetler de CM’lerde yaygındır. (Bu hastalık hakkında daha fazla bilgi için Nadir Hastalık Veritabanında arama teriminiz olarak “kavernöz hemanjiyom”u seçin.)
Venöz anjiyomlar (VA’lar), boyutları değişen ancak arterleri içermeyen genişlemiş, dolaşık ve bükülmüş damarları içerir. Bu “büyümelerin” yeri, çoğunlukla damarın kılcal aşamasının hemen sonrasıdır (kapiller sonrası malformasyon). İzole kusurlar veya kavernöz malformasyonlarla ilişkili olabilirler. Kusur, daha büyük bir damarın (gövde) bir parçasını oluşturmak için bir araya gelen küçük damarların (venüller) bir “tacı” olarak kendini gösterir.
Telenjiektaziler, ince kılcal damarların genişlemesi (genişlemesi) sonucu ortaya çıkan malformasyonlardır. Bu genişlemiş kılcal damarlar, yüz, gözler, beyni (dura) ve omuriliği (meninksler) kaplayan zarlar ve mukoza zarları (beyni kaplayan ince nemli tabaka) gibi vücudun çeşitli yerlerinde küçük pembe-kırmızı noktalar olarak bilinirler. vücudun iç yüzeyleri). (Telanjiektazileri içeren bir hastalık hakkında daha fazla bilgi için, Nadir Hastalık Veritabanında arama teriminiz için “hemorajik telenjiektazi, kalıtsal”ı seçin.)
Galen damarı malformasyonları (VGM’ler embriyo gelişirken başlar. Galen damarı serebral hemisferlerin altında bulunur ve beynin ileri (anterior) ve merkezi bölgelerini uygun sinüslere boşaltır. Malformasyonlar, Galen damarı bozulduğunda ortaya çıkar. kafa içinde çevre doku tarafından desteklenmez ve normal fibröz duvardan yoksundur. Böylece, Galen damarı serebral boşlukların (sinüsler) sıvıları içinde serbest yüzer gibi görünür. Galen damarı içindeki basınç artarsa, şekli değişir. silindirden küreye. Bu tür değişikliklere anormal fetal kan dolaşımı eşlik eder. Aşırı durumlarda, kalp yetmezliği veya beyinde şişme (hidrosefali) olabilir.
Karışık malformasyon, birkaç çoklu karışık malformasyondan herhangi birini dahil etmek için kullanılan bir ifadedir. Sıklıkla, bu malformasyonlar, arteriyovenöz malformasyonların telenjiektazilerle karışımı gibi görünmektedir.
Nedenleri
VMB’nin üç tipi veya formu genetik bir bileşene sahiptir. Kavernöz hemanjiyomlar ve telenjiektazilerde genetik bir neden olduğuna dair kanıtlar güçlüdür. Beynin arteriovenöz malformasyonu (AVM) için durum çok daha zayıftır. Bu vakaların her birinde, durum otozomal dominant bir özellik olarak iletilir. Kavernöz malformasyonlar durumunda arızalı gen, gen haritası lokusu 7q11.2-q21’e ve telenjiektazi durumunda gen haritası lokusu 9q34.1’e kadar izlendi.
İnsan hücrelerinin çekirdeğinde bulunan kromozomlar, her bireyin genetik bilgisini taşır. İnsan vücut hücreleri normalde 46 kromozoma sahiptir. İnsan kromozom çiftleri 1’den 22’ye kadar numaralandırılır ve cinsiyet kromozomları X ve Y olarak adlandırılır. Erkeklerde bir X ve bir Y kromozomu bulunurken dişilerde iki X kromozomu bulunur. Her kromozomun “p” olarak adlandırılan kısa bir kolu ve “q” olarak adlandırılan uzun bir kolu vardır. Kromozomlar ayrıca numaralandırılmış birçok banda bölünmüştür. Örneğin, “kromozom 9q34.1”, 7. kromozomun uzun kolundaki 34.1 bandına karşılık gelir. Numaralandırılmış bantlar, her bir kromozomda bulunan binlerce genin konumunu belirtir.
Anormal genin 7q11.2-q21’de bulunduğunu söylemek, söz konusu genin 7. kromozomun uzun kolunda 11.2 ve 21. bantlar arasındaki bir bölgede yer aldığı anlamına gelir.
Genetik hastalıklar, anne ve babadan alınan kromozomlar üzerinde bulunan belirli bir özellik için genlerin birleşmesi ile belirlenir. Baskın genetik bozukluklar, hastalığın ortaya çıkması için anormal bir genin yalnızca tek bir kopyası gerekli olduğunda ortaya çıkar. Anormal gen, her iki ebeveynden de kalıtsal olabilir veya etkilenen bireyde yeni bir mutasyonun (gen değişikliği) sonucu olabilir. Anormal genin etkilenen ebeveynden yavruya geçme riski, ortaya çıkan çocuğun cinsiyetine bakılmaksızın her gebelik için %50’dir.
Resesif genetik bozukluklar, bir birey, her bir ebeveynden aynı özellik için aynı anormal geni miras aldığında ortaya çıkar. Bir birey, hastalık için bir normal gen ve bir gen alırsa, kişi hastalığın taşıyıcısı olacak, ancak genellikle semptom göstermeyecektir. Taşıyıcı iki ebeveynin her ikisinin de kusurlu geni geçirme ve dolayısıyla etkilenen bir çocuğa sahip olma riski her hamilelikte %25’tir. Anne baba gibi taşıyıcı olan bir çocuğa sahip olma riski her gebelikte %50’dir. Bir çocuğun her iki ebeveynden de normal genler alma ve bu özel özellik için genetik olarak normal olma şansı %25’tir. Risk erkekler ve kadınlar için aynıdır.
Tüm bireyler birkaç anormal gen taşır. Yakın akraba (akraba) olan ebeveynlerin her ikisinin de aynı anormal geni taşıma şansı akraba olmayan ebeveynlere göre daha yüksektir, bu da resesif genetik bozukluğu olan çocuklara sahip olma riskini artırır.
X’e bağlı resesif genetik bozukluklar, X kromozomundaki anormal bir genin neden olduğu durumlardır. Dişilerde iki X kromozomu vardır ancak X kromozomlarından biri “kapalı” durumdadır ve o kromozom üzerindeki tüm genler inaktive edilmiştir. X kromozomlarından birinde hastalık geni bulunan dişiler, bu bozukluğun taşıyıcılarıdır. Taşıyıcı dişiler genellikle bozukluğun semptomlarını göstermezler çünkü genellikle “kapalı” olan anormal gene sahip X kromozomudur. Bir erkeğin bir X kromozomu vardır ve eğer bir hastalık geni içeren bir X kromozomunu miras alırsa, hastalığı geliştirecektir. X’e bağlı bozuklukları olan erkekler, hastalık genini taşıyıcı olacak tüm kızlarına aktarır. Bir erkek, X’e bağlı bir geni oğullarına geçiremez çünkü erkekler, erkek yavrulara her zaman X kromozomları yerine Y kromozomlarını aktarır.
X’e bağlı baskın bozukluklara ayrıca X kromozomundaki anormal bir gen neden olur, ancak bu nadir durumlarda, anormal gene sahip dişiler hastalıktan etkilenir. Anormal gene sahip erkekler, kadınlardan daha ciddi şekilde etkilenir ve bu erkeklerin çoğu hayatta kalamaz.
Teşhisi
Manyetik rezonans görüntüleme (MRI), bilgisayarlı tomografi (BT) taramaları, venogramlar ve/veya dijital intravenöz veya genel anjiyografi gibi görüntüleme cihazları, vasküler malformasyonların olup olmadığını görmek için beyin kan damarlarının resimlerini çekebilir.
Tedavisi
Mevcut tedavi seçenekleri, malformasyonun ciddiyetine ve yerine göre değişir. Cerrahi çıkarma (rezeksiyon), çoklu embolizasyon (anormal kan damarlarına giden ve/veya bu damarlardan kan akışını engellemek için dolaşım sistemine peletlerin konulduğu bir ameliyat) ve ışınlama şu anda kullanılan tedavilerdir. Bazı durumlarda tedavi gerekmeyebilir. Son zamanlarda tanıtılan teknikler, parçacık ışını ve stereotaksik radyo-cerrahiyi içerir. Genetik danışmanlık, bu bozukluğun kalıtsal bir formuna sahiplerse hastalar ve aileleri için faydalı olabilir. Diğer tedavi semptomatik ve destekleyicidir.
Not: Sunulan bilgilerin amacı herhangi bir hastalığı teşhis veya tedavi etmek, iyileştirmek veya önlemek değildir. Tüm bilgiler yalnızca genel bilginize yöneliktir, tıbbi tavsiye veya belirli tıbbi durumların tedavisinin yerine geçmez. Uygulamadan önce bu bilgileri doktorunuzla görüşün.