XP, otozomal resesif bir genetik bozukluktur. Resesif genetik bozukluklar, bir birey aynı özellik için bir genin çalışmayan iki kopyasını miras aldığında ortaya çıkar. Bir gen anneden, diğeri babadan gelir. Bir birey, durum için bir çalışan gen ve bir çalışmayan gen miras alırsa, kişi durum için bir taşıyıcı olacaktır, ancak genellikle semptom göstermeyecektir. Taşıyıcı iki ebeveynin hem çalışmayan geni geçirme hem de etkilenen bir çocuğa sahip olma riski her hamilelikte %25’tir. Anne baba gibi taşıyıcı çocuk sahibi olma riski her gebelikte %50’dir. Bir çocuğun her iki ebeveynden de çalışma genleri alma şansı %25’tir. Hastalığı kalıtsal olarak alma riski kadın ve erkekler için aynıdır.
Kan akrabası (akraba) olan ebeveynlerin, akraba olmayan ebeveynlere göre her ikisinin de aynı çalışmayan geni taşıma şansı daha yüksektir, bu da resesif genetik bozukluğu olan çocuklara sahip olma riskini artırır.
XP hastalarında çalışmayan 9 farklı gen vardır ve şunları içerir: DDB2 (XP-E), ERCC1 , ERCC2 (XP-D), ERCC3 (XP-G), ERCC4 (XP-F), ERCC5 (XP-B), POLH (XP-V veya varyantı) , XPA ve XPC .
Bu genlerin normal ifadesinden kaynaklanan proteinler, DNA onarımında yer alır ve hasarlı DNA’yı tanımaya, hasarı gidermeye ve oluşan boşluğu doldurmaya yarar. Bir kişi bir çift çalışmayan XP genini miras aldığında, UV’den kaynaklanan hasarı düzgün bir şekilde onaramaz ve durumun belirtilerini gösterir.

Tedavisi
Devam eden DNA hasarını ve hastalığın ilerlemesini önlemek için tanıdan şüphelenildiği anda başlamak üzere sıkı bir güneş (UV) koruması gereklidir. XP’li bireyler, cildi ve gözleri ultraviyole (UV) radyasyona maruz bırakmaktan kaçınmalıdır. Bu, şapkalar, UV’yi engelleyen yüz siperlikleri olan başlıklar, uzun kollu giysiler, pantolonlar ve eldivenler gibi koruyucu giysiler giyilerek yapılabilir. Yüksek güneş koruyucu faktörlü (SPF) güneş kremleri, yan siperleri olan UV’den koruyan gözlükler ve uzun saçlar da koruma sağlayabilir.
XP hastasının çevresi (örn. ev, okul ve iş) bir UV ışık ölçer kullanılarak UV seviyeleri açısından test edilmelidir. Ölçüm cihazı, artan UV alanlarını ve zararlı UV kaynaklarını (örn. halojen ve korumasız floresan ampuller ve cıva buharlı lambalar) belirlemeye yardımcı olabilir. Bu UV kaynakları ortamdan uzaklaştırılmalıdır. UV camdan geçebildiğinden, XP hastasının evlerinde, okullarında, iş yerlerinde ve arabalarında bulunan dul kadınlara UV engelleyici film ile tedavi edilmelidir.
D vitamini, sağlıklı kemiklerin korunmasına yardımcı olan temel bir vitamindir. D vitamini, UV’nin cilt ile etkileşimi ile üretilir. XP’li kişiler UV’den kaçındığından, yetersiz D vitamini seviyelerinin komplikasyonlarını önlemek için gerektiğinde oral diyet takviyeleri alınabilir.
Sigara dumanındaki bazı kanserojenler UV’ye benzer şekillerde DNA’ya zarar verir ve ikinci el sigara dumanına maruz kalmaktan kaçınılmalıdır. Sigara içen XP hastalarında akciğer kanseri gelişmiştir.
Cilt (kafa derisi, dudaklar, dil ve göz kapakları dahil) kanser öncesi ve kanserli lezyonları saptamak için her 6-12 ayda bir (veya gerekirse daha sık) bir dermatolog tarafından muayene edilmelidir. Lezyonların daha fazla büyümesini veya yayılmasını önlemek için herhangi bir cilt kanserinin derhal çıkarılması gereklidir. Etkilenen bireyler ve çocukların vasileri, olası cilt kanserlerinin erken teşhisine yardımcı olmak için cilt muayenesi teknikleri konusunda bilgilendirilmelidir.
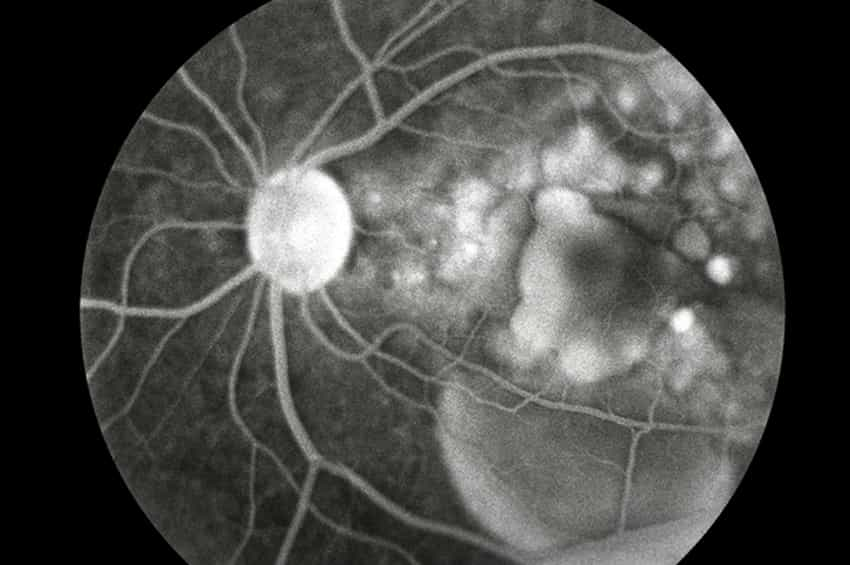
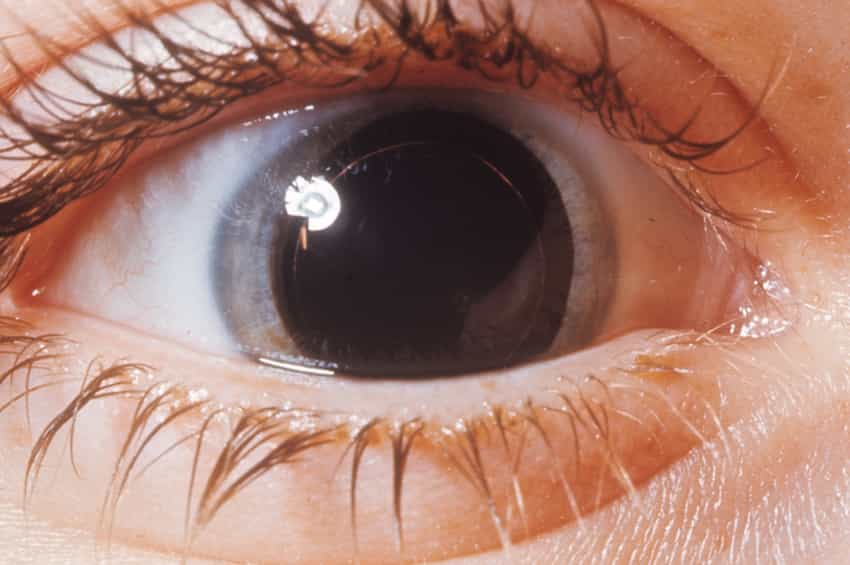
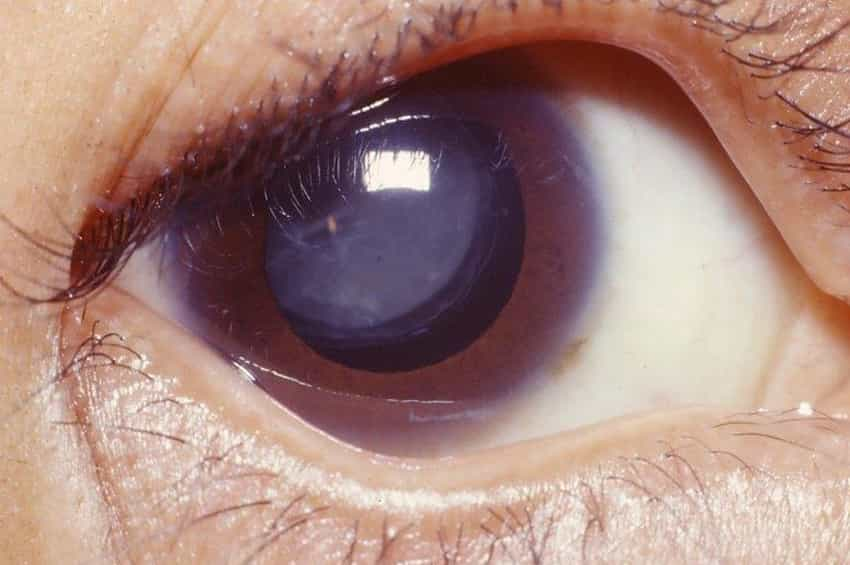
Bireyler ayrıca bir göz doktoru tarafından rutin göz muayenelerinden geçmelidir. Göz kapakları ektropion (sarkma ve sarkma), entropion (göz tahrişine neden olabilen içe dönme) ve pterygia/pinguecula (göz yüzeyindeki iyi huylu büyümeler) açısından incelenmelidir. Gözü örten kornea bulanıklık açısından değerlendirilmeli ve gözler kuruluk açısından test edilmelidir; Schirmer testinde gözyaşı emilimini ölçmek için göz kapaklarının altına bir filtre kağıdı yerleştirilir. Retinadaki (gözün arkası) herhangi bir değişikliği değerlendirmek için genişlemiş bir göz muayenesi önemlidir.
Oksipital frontal çevrenin ölçülmesi (mikrosefali varlığını belirlemek için) ve derin tendon reflekslerinin varlığının değerlendirilmesini içeren temel nörolojik muayeneler, bir XP hastasının rutin bakımının bir parçası olmalıdır. Nörolojik hastalığı olan XP’nin bir göstergesi olan erken başlangıçlı işitme kaybını değerlendirmek için düzenli olarak işitme muayeneleri yapılmalıdır. İşitme kaybı tespit edilirse, işitme cihazları eksikliklerin düzeltilmesinde çok faydalı olabilir. Nörolojik problemler tespit edilirse, bir nörolog tarafından daha derinlemesine incelemeler yapılır. Ek olarak, MRG’ler, nörodejenerasyonu olan XP hastalarının beyinlerinde yaygın olarak görülen değişiklikleri değerlendirebilir. Nörolojik hastalık geliştiren XP hastalarında periferik nöropati olabilir,
Küçük, premalign cilt lezyonları (örn. aktinik keratoz), sıvı nitrojen ile dondurularak tedavi edilebilir. Hasarlı derinin daha geniş alanları için 5-florourasil veya imikimod gibi topikal kremler uygulanabilir. Dermatom tıraşı ve dermabrazyon gibi cerrahi prosedürler daha geniş cilt alanları için kullanılmıştır. Gövde ve ekstremitelerdeki küçük deri kanserleri, elektrodezikasyon ve küretaj veya cerrahi eksizyon ile tedavi edilebilir. Derin invaziv cilt kanserleri veya yüz ve doku koruyucu teknikler gerektiren bölgelerdeki cilt kanserleri Mohs mikrografik cerrahi ile tedavi edilebilir.
Şiddetli vakalarda, cildin büyük bölümleri güneşten korunan deri ile yeniden aşılanabilir (veya değiştirilebilir). X-ışını tedavisi, ameliyat edilemeyen veya daha büyük neoplazmları tedavi etmek için veya cerrahiye adjuvan tedavi olarak kullanılabilir. Çoklu bazal hücreli karsinomlar, oral vismodegib ile tedavi edilebilir. bir sonik kirpi yolu inhibitörü. Son zamanlarda, büyük deri tümörlerini ve metastatik kanserleri tedavi etmek için kontrol noktası inhibitörleri olarak adlandırılan yeni kemoterapi ajanları kullanılmıştır. Tümörleri tedavi etmede başarılı oldular; ancak ciddi yan etkiler görülebilir.
Oral retinoidler izotretinoin veya asitretin, yeni cilt neoplazmalarını önlemek için kullanılabilir, ancak karaciğer toksisitesi, yüksek kolesterol seviyeleri, bağ ve tendonların kalsifikasyonu ve büyüyen kemik şaftlarının erken kapanması gibi birçok yan etkiye sahiptir. Bu retinoid ilaçların doğum kusurlarına neden olduğu bilinmektedir ve hamile kadınlarda veya hamile kalmaya çalışan kadınlarda kontrendikedir. Tümörleri tedavi etmede başarılı oldular; ancak ciddi yan etkiler görülebilir. Oral retinoidler izotretinoin veya asitretin, yeni cilt neoplazmalarını önlemek için kullanılabilir, ancak karaciğer toksisitesi, yüksek kolesterol seviyeleri, bağ ve tendonların kalsifikasyonu ve büyüyen kemik şaftlarının erken kapanması gibi birçok yan etkiye sahiptir.
Bu retinoid ilaçların doğum kusurlarına neden olduğu bilinmektedir ve hamile kadınlarda veya hamile kalmaya çalışan kadınlarda kontrendikedir. Tümörleri tedavi etmede başarılı oldular; ancak ciddi yan etkiler görülebilir. Oral retinoidler izotretinoin veya asitretin, yeni cilt neoplazmalarını önlemek için kullanılabilir, ancak karaciğer toksisitesi, yüksek kolesterol seviyeleri, bağ ve tendonların kalsifikasyonu ve büyüyen kemik şaftlarının erken kapanması gibi birçok yan etkiye sahiptir. Bu retinoid ilaçların doğum kusurlarına neden olduğu bilinmektedir ve hamile kadınlarda veya hamile kalmaya çalışan kadınlarda kontrendikedir.
Sık kullanılan kayganlaştırıcı göz damlaları korneayı nemli tutar ve kuru gözün iltihabik etkilerine karşı korur. Deforme göz kapaklarının neden olduğu mekanik travmaya karşı korunmak için yumuşak kontakt lensler takılabilir. Önce daha basit tedavilerle başlamak en iyisidir.
Göz kapakları, konjonktiva ve kornea neoplazmaları ameliyatla tedavi edilebilir. Bazı durumlarda, UV kaynaklı oküler hasarı ve kornea bulanıklığını düzeltmek için kornea nakli denenmiştir. Bununla birlikte, bağışıklığın reddi nedeniyle nakiller başarılı olmayabilir. Ne yazık ki, bağışıklık reddini önlemek için kullanılan bağışıklık baskılayıcı ilaçlar ek cilt kanserlerine yol açabilir. Topikal kemoterapi ajanları, gözün konjonktiva ve sklerasındaki tümörleri tedavi etmek için kullanılabilir.
Nörolojik anormallikler, artan yüksek frekanslı duyusal-nöral işitme kaybıyla ilişkilidir. İşitme kaybı ilerleyicidir (zamanla kötüleşir) ve işitme cihazlarıyla tedavi edilebilir. Son zamanlarda, bazı XP hastalarında koklear implantlar kullanılmıştır. Çocuklukta ve özel eğitim sınıflarında bilişsel gecikmeler görülebilir, fiziksel ve mesleki terapiler ile okuldaki UV korumalı konaklamalar XP çocukları için çok faydalıdır. Yaşlandıkça, XP nörolojik hastalığı olan kişiler, durum ilerledikçe artan ataksi, disfaji (yutma güçlüğü) ve dizartri (konuşma güçlüğü) yaşarlar. Tekerlekli sandalyeye, beslenme tüplerine ve uzun süreli hemşirelik bakımına ihtiyaç duyabilirler.
Not: Sunulan bilgilerin amacı herhangi bir hastalığı teşhis veya tedavi etmek, iyileştirmek veya önlemek değildir. Tüm bilgiler yalnızca genel bilginize yöneliktir, tıbbi tavsiye veya belirli tıbbi durumların tedavisinin yerine geçmez. Uygulamadan önce bu bilgileri doktorunuzla görüşün.