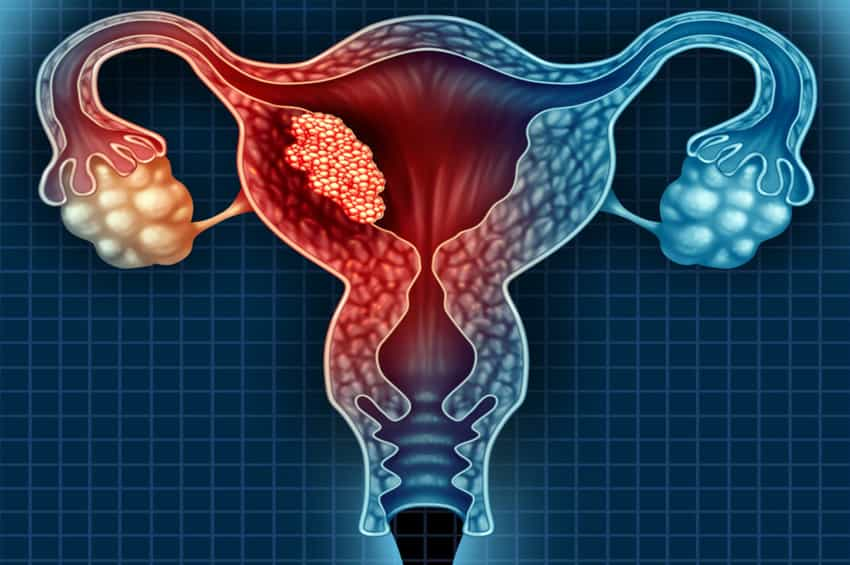
Rahim (uterin) leiomyosarkomu, rahmin duvarlarını (miyometrium) kaplayan düz kastan kaynaklanan ve nadir görülen bir malign (kanserli) tümördür. Vücutta temelde iki tür kas vardır: istemli ve istemsiz. Düz kaslar istemsiz kaslardır; beynin onlar üzerinde bilinçli bir kontrolü yoktur. Düz kaslar, çeşitli uyaranlara yanıt olarak istemsiz olarak tepki verir.
Haber Merkezi / Örneğin, miyometriyum hamilelik sırasında fetüsün uyum sağlamasına yardımcı olmak için gerilir ve doğum sırasında bebeğin dışarı itilmesine yardımcı olmak için kasılır. Leiomyosarkom bir kanser türüdür. “Kanser” terimi, çevreleyen dokuları istila eden ve kan dolaşımı, lenfatik sistem veya başka yollarla uzak vücut dokularına veya organlarına yayılabilen (metastaz yapabilen) anormal, kontrolsüz hücresel büyüme ile karakterize edilen bir hastalık grubunu ifade eder.
Farklı kanser türleri, leiomyosarkomlar dahil olmak üzere, ilgili hücre tipine, malignitenin spesifik doğasına ve hastalığın klinik seyrine göre sınıflandırılabilir. Leiomyosarkom yumuşak doku sarkomu olarak sınıflandırılır. Sarkomlar, vücuttaki çeşitli yapı ve organları birbirine bağlayan, destekleyen ve çevreleyen bağ dokusundan kaynaklanan kötü huylu tümörlerdir. Yumuşak doku, yağ, kas, asla, tendonlar, eklemleri çevreleyen doku (sinovyal doku), kan ve lenf damarlarını içerir.
Rahim leiomyosarkom da dahil olmak üzere leiomyosarkomun kesin nedeni bilinmemektedir. vücuttaki çeşitli yapı ve organları destekler ve çevreler. Yumuşak doku, yağ, kas, tendonlar, eklemleri çevreleyen doku (sinovyal doku), kan ve lenf damarlarını içerir. Rahim leiomyosarkom da dahil olmak üzere leiomyosarkomun kesin nedeni bilinmemektedir. vücuttaki çeşitli yapı ve organları destekler ve çevreler. Yumuşak doku, yağ, kas, asla, tendonlar, eklemleri çevreleyen doku (sinovyal doku), kan ve lenf damarlarını içerir.
Belirtileri ve semptomları
Rahim leiomyosarkomunun semptomları, tümörün yeri, boyutu ve ilerlemesine bağlı olarak vakadan vakaya değişebilir. Pek çok kadının herhangi bir belirgin semptom göstermeyebilir. En yaygın semptom vajina ve uterustan anormal kanamadır.
Pelvis veya mideyi etkileyen basınç veya ağrı, anormal vajinal akıntı ve mesane veya bağırsak alışkanlıklarında değişiklik gibi ek semptomlar ortaya çıkabilir. Genellikle kanserle ilişkilendirilen genel semptomlar arasında yorgunluk, ateş, kilo kaybı ve genel bir sağlıksızlık hissi (halsizlik) yer alır.
Uterin leyomiyosarkomlar kötü huyludur ve lokal olarak ve vücudun diğer bölgelerine, özellikle akciğerler ve karaciğere yayılabilir (metastaz yapabilir) ve sıklıkla yaşamı tehdit eden komplikasyonlara neden olur. Leiomyosarkomlar vakaların yarısından fazlasında bazen ilk tanı ve tedaviden sonra sekiz ila 16 ay içinde tekrarlar.
Nedenleri
Rahim leiomyosarkomunun kesin nedeni bilinmemektedir. Genellikle görünürde bir sebep olmaksızın kendiliğinden ortaya çıkarlar. Son derece nadir durumlarda, bazı araştırmacılar iyi huylu (kanserli olmayan) bir fibroidde kötü huylu bir değişikliği (dönüşüm) işaret edebileceklerine inanmaktadır.
Bununla birlikte, diğer araştırmacılar, fibroidlerin habis dönüşümünün kanıtlanmadığını iddia etmektedir. Miyomlar kadınları etkileyen en yaygın tümörlerdir ve sıklıkla çocuk doğurma yıllarında ortaya çıkarlar. Miyomlar ayrıca leiomyomlar veya miyomlar olarak da bilinebilir. Miyomların çoğu herhangi bir semptom veya komplikasyona neden olmaz.
Araştırmacılar, genetik ve immünolojik anormalliklerin, çevresel faktörlerin (örn. ultraviyole ışınlarına, belirli kimyasallara, iyonlaştırıcı radyasyona maruz kalma), diyetin, stresin ve/veya diğer faktörlerin belirli kanser türlerinin oluşumunda rol oynayabileceğini düşünüyorlar.
Leiomyosarkom da dahil olmak üzere kanserli bireylerde, onkogenler veya tümör baskılayıcı genler olarak bilinen belirli hücrelerin yapısındaki ve yönelimindeki anormal değişiklikler nedeniyle maligniteler gelişebilir. Onkogenler hücre büyümesini kontrol eder; tümör baskılayıcı genler, hücre bölünmesini kontrol eder ve hücrelerin uygun zamanda ölmesini sağlar.
Bu genlerdeki değişikliklerin spesifik nedeni bilinmemektedir. Bununla birlikte, mevcut araştırmalar, vücudun genetik kodunun taşıyıcısı olan DNA’daki (deoksiribonükleik asit) anormalliklerin, hücresel malign transformasyonun altında yatan temel olduğunu düşündürmektedir. Bu anormal genetik değişiklikler, bilinmeyen nedenlerle kendiliğinden ortaya çıkabilir veya daha nadiren kalıtsal olabilir.
Leiomyosarkomlar spesifik genetik ve çevresel risk faktörleri ile ilişkilidir. Aileden geçen bazı kalıtsal durumlar leiomyosarkom gelişme riskini artırabilir. Bu bozukluklar arasında Gardner sendromu, Li-Fraumeni sendromu, Werner sendromu, nörofibromatozis ve birkaç bağışıklık yetersizliği sendromu yer alır. Leiomyosarkom ile bu bozukluklar arasındaki kesin ilişki anlaşılamamıştır.
Teşhisi
Rahim leiomyosarkom tanısı, en sık olarak, etkilenen bireyler uterusun iyi huylu düz kas tümörleri (leiomyomlar veya fibroidler) için ameliyat edildiğinde tesadüfen konur. Miyom cerrahisi örneklerinin patolojik incelemesi, mikroskop altında iyi huylu bir miyomdan çok bir leiomyosarkomun karakteristiği olan belirli özellikleri ortaya çıkarabilir. Bu nedenle, patolojik inceleme, malign leiomyosarkomu iyi huylu muadili olan leiomyomadan ayırt eden anahtar tanısal yönüdür.
Tümörün boyutunu, yerleşimini ve yayılımını değerlendirmeye yardımcı olmak ve uterin leiomyosarkomlu bireylerde gelecekteki cerrahi prosedürlere yardımcı olmak için özel görüntüleme teknikleri kullanılabilir. Bu tür görüntüleme teknikleri arasında bilgisayarlı tomografi (CT) taraması, manyetik rezonans görüntüleme (MRI) ve ultrason yer alabilir.
BT taraması sırasında, belirli doku yapılarının enine kesit görüntülerini gösteren bir film oluşturmak için bir bilgisayar ve röntgen ışınları kullanılır. MRI, belirli organların ve vücut dokularının enine kesit görüntülerini üretmek için bir manyetik alan ve radyo dalgaları kullanır. Ultrason sırasında yansıyan ses dalgaları uterusun bir görüntüsünü oluşturur. Bölgesel lenf düğümlerinin olası infiltrasyonunu ve uzak metastazların varlığını belirlemek için laboratuvar testleri ve özel görüntüleme testleri de yapılabilir.
Metastatik leiomyosarkom tanısını doğrulamak için ince iğne aspirasyonu yapılabilir. İnce ihtiyaç aspirasyonu (İİA), ince, içi boş bir iğnenin deriden geçirildiği ve küçük doku örnekleri almak için nodül veya kitleye sokulduğu bir teşhis tekniğidir. Toplanan doku daha sonra mikroskop altında incelenir. Bazı durumlarda, İİA sonuçsuz kalabilir ve doktorlar insizyonel biyopsi yapabilir. İnsizyonel biyopsi sırasında, küçük bir örnek doku cerrahi olarak çıkarılır ve mikroskobik yapısını ve yapısını (histopatoloji) belirlemek için işlendiği ve incelendiği bir patoloji laboratuvarına gönderilir.
Tedavisi
Spesifik prosedürler ve müdahaleler, birincil tümörün konumu, birincil tümörün kapsamı (evre) ve malignite derecesi (derecesi) gibi çok sayıda faktöre bağlı olarak değişebilir; tümörün lenf düğümlerine veya uzak bölgelere yayılıp yayılmadığı; bireyin yaşı ve genel sağlığı; ve/veya diğer unsurlar. Belirli müdahalelerin kullanılmasına ilişkin kararlar, hekimler ve sağlık ekibinin diğer üyeleri tarafından, vakasının özelliklerine göre hastayla dikkatli bir şekilde istişare edilerek verilmelidir; potansiyel faydalar ve risklerin kapsamlı bir şekilde tartışılması; hasta tercihi; ve diğer uygun faktörler.
Rahim leiomyosarkomu için birincil tedavi şekli, tüm tümörün ve etkilenen herhangi bir dokunun cerrahi olarak çıkarılmasıdır. Rahmin tamamen cerrahi olarak çıkarılması (histerektomi) genellikle yapılır. Özellikle menopoza giren veya metastazı olan kadınlar için fallop tüplerinin ve yumurtalıkların cerrahi olarak çıkarılması (bilateral salpingo-ooferektomi) önerilebilir.
Tek başına veya kombinasyon halinde (kemoterapi) ve radyasyon tedavisi olarak kullanılan anti-kanser ilaçları, cerrahi (adjuvan tedavi) sonrasında leiomyosarkomlu bireyleri sınırlı fayda ile tedavi etmek için kullanılmıştır. Leiomyosarkomların tedavisinde faydalı olabilecek yeni kemoterapötik kombinasyonlar geliştirmek için araştırma terapileri devam etmektedir. Bazı durumlarda, bir tümörün boyutunu küçültmek için ameliyattan önce radyasyon tedavisi kullanılabilir.
Leiomyosarkomların tekrarlama riski yüksek olduğu için bireyler ameliyattan sonra rutin olarak muayene edilmelidir.
Not: Sunulan bilgilerin amacı herhangi bir hastalığı teşhis veya tedavi etmek, iyileştirmek veya önlemek değildir. Tüm bilgiler yalnızca genel bilginize yöneliktir, tıbbi tavsiye veya belirli tıbbi durumların tedavisinin yerine geçmez. Uygulamadan önce bu bilgileri doktorunuzla görüşün.